
Ätiologisch, pathologisch und klinisch uneinheitliche Nephropathie
Entzündliche Erkrankungen der Glomeruli können durch Schädigung aller in den Filtereinheiten vertretenen
Zellen und Strukturen verursacht werden. Mit wenigen Ausnahmen handelt es sich bei der Glomerulonephritis (GN)
um eine immunvermittelte Erkrankung, in deren Pathophysiologie neben Antikörpern auch Chemokine, Zytokine,
Wachstumsfaktoren, Komplementaktivierung, Koagulationsprodukte, Leukozyten und die Nierenzellen selbst eine
Rolle spielen. Ätiologisch werden primäre Nierenerkrankungen aufgrund akut entzündlicher Glomerulopathien,
akut entzündliche Glomerulonephritiden als Begleitreaktion bei Infektionen und Begleitreaktion systemischer
Erkrankungen unterschieden. Der Behandlung von Glomerulonephritiden ist in zahlreichen Fällen kein Erfolg
beschieden, so daß die Erkrankung eine der häufigsten Ursachen für terminales Nierenversagen ist.
Klinische Leitsymptome aussagekräftiger als pathologische Charakteristika
-
In die klassische Einteilung der Glomerulonephritiden fließen ätiologische, histopathologische und
klinische Gesichtspunkte ein. Diese Durchmischung ist für den praktisch tätigen Arzt oft nicht sehr
hilfreich, denn das klinische Erscheinungsbild kann vielfach auf verschiedene GN-Formen hinweisen.
Insbesondere wird zwischen der Ausbildung eines nephrotischen Syndroms und einem (akut) nephritischen Verlauf

Das klinische Erscheinungsbild der Glomerulonephritiden kann sehr variabel sein:
Asymptomatische Verläufe kommen zufällig bei Urinuntersuchungen zum Vorschein. Man findet unter Umständen
eine milde Proteinurie und/oder eine Mikrohämaturie, vielfach ohne daß der Patient Ödeme aufweist und einen
Bluthochdruck entwickelt hat.
Der Patient mit einem nephritischen Syndrom präsentiert sich mit einer erst kurz zuvor eingetretenen
Hämaturie und Proteinurie. Die Nierenfunktion ist beeinträchtigt und durch Wasser- und Salzretention
entsteht Bluthochdruck. Das nephritische Krankheitsbild herrscht bei der rapid progressiven GN und der
akuten postinfektiösen GN vor. Teilweise wird es aber auch bei der membranoproliferativen und der
mesangioproliferativen GN beobachtet.
Bei der rapid progressiven GN kommt es innerhalb von Tagen bis drei Monaten zu einem Abfall der
glomerulären Filtrationsrate um mindestens 50 % bei zumeist nephritischem Krankheitsbild und ausgeprägter
Halbmondbildung. Als Verlaufsform kann die rapid progressive GN bei praktisch allen GN-Typen auftreten.
Die Bezeichnung geht auf einige Fälle von poststreptokokkaler GN mit ungewöhnlich fulminantem Verlauf
zurück.
Das nephrotische Syndrom ist durch eine erhebliche Proteinurie (>3,5 g/1,73 m2/Tag), Hypalbuminämie,
Hyperlipidämie und Ödeme gekennzeichnet.
Bei der chronischen GN nimmt die Erkrankung einen schleichenden Verlauf. Der Beginn ist meist nicht
feststellbar. Über Jahre und Jahrzehnte entwickelt sich vielfach eine Niereninsuffizienz. Histopathologisch
liegt der chronischen GN in den meisten Fällen eine mesangioproliferative GN, eine membranoproliferative GN
oder eine fokal sklerosierende GN zugrunde.
Glomerulonephritiden treten beim männlichen Geschlecht deutlich häufiger auf als beim weiblichen. Die Ausnahme
ist Lupus nephritis. Aber selbst da zeigt sich die verstärkte Anfälligkeit der Männer für Glomerulonephritiden,
denn der Geschlechterunterschied ist erheblich geringer ausgeprägt als beim systemischen Lupus erythematodes.
|
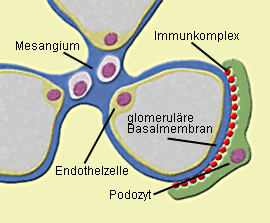
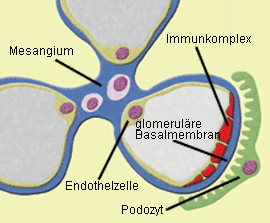
Abb. 1a: Membranöse GN: Zwischen glomerulärer Basalmembran und Podozyten lagern sich
Immunkomplexe in granulärer Form ab, so daß es zu einer Schädigung der Podozytenfortsätze kommt. 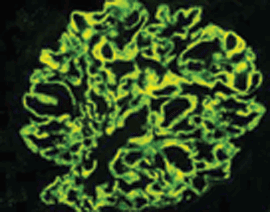 Abb.1b: Membranöse GN (IgG-Immunfluoreszenz): Die glomeruläre Basalmembran erscheint
als diffuses granuläres Muster.
Abb.1b: Membranöse GN (IgG-Immunfluoreszenz): Die glomeruläre Basalmembran erscheint
als diffuses granuläres Muster.
|
-
Die Proteinurie beim nephrotischen Syndrom ist auf die erhöhte Permeabilität des Harnfilters und nicht auf
verminderte tubuläre Resorption zurückzuführen. Neben vorherrschend Albumin gehen auch Immunglobulin G und
Gerinnungsfaktoren verloren. Als pathologisches Merkmal findet man auf der Harnseite der Glomeruli eine
Schädigung der Podozyten.
Die Minimalläsion (Minimal Change Disease) ist die häufigste Ursache für ein nephrotisches
Syndrom im Kindesalter. In den meisten Fällen spricht die Krankheit bei Kindern gut auf eine Steroidtherapie
an und ist reversibel.
Bei 20 bis 30% der Erwachsenen mit einem nephrotischen Syndrom läßt sich eine Minimalläsion nachweisen.
Die Krankheit ist gegenüber einer Behandlung mit Steroiden vielfach resistent, oder es kommt zum Rückfall.
In solchen Fällen kann unter Umständen mit Cyclosporin A und Cyclophosphamid erfolgreich behandelt werden.
Die Minimalläsion zeigt lichtmikroskopisch keine Auffälligkeiten. Erst im Elektronenmikroskop läßt sich die
Verschmelzung der Podozytenfortsätze erkennen.
Die membranöse GN ist die häufigste Ursache eines nephrotischen Syndroms im Erwachsenenalter.
Zur Schädigung des Harnfilters kommt es durch Ablagerung von Immunkomplexen zwischen den Podozytenfortsätzen
und der glomerulären Basalmembran (Abb. 1a, b). Sofern die Ursache hierfür nicht bekannt ist, wird von einer
primären (idiopathischen) Erkrankung gesprochen. Vielfach tritt die membranöse GN jedoch im Rahmen anderer
Erkrankungen wie dem systemischen Lupus erythematodes oder einer Infektion mit Hepatisis-B-Viren auf.
Ursächlich kann auch die Einnahme von Medikamenten wie Captopril, Penicillamin sowie von Gold- und
Quecksilberpräparaten sein. Ferner kommt die membranöse GN bei Krebserkrankungen wie insbesondere Bronchial-,
Kolon- und Prostatakarzinom vor.
In etwa jedem vierten Fall von membranöser GN kommt es zu einer dauerhaften Spontanremission. Bei einem ähnlich
hohen Anteil der Patienten entwickelt sich eine unterschiedlich stark ausgeprägte persistierende Proteinurie mit
nichturämischer Azotämie. In etwa jedem zweiten Fall schreitet die Krankheit bis zur terminalen Niereninsuffizienz
voran.
Für die Therapie der idiopathischen membranösen GN sind Steroide allein völlig unwirksam. Vielmehr muß Prednisolon
mit Chlorambucil, Cyclophosphamid oder Cyclosporin A kombiniert werden. Bei günstigen prognostischen Voraussetzungen
(Proteinurie <6 g/d und Kreatinin <1,5 mg/dl) ist es aufgrund der hohen Rate an Spontanremissionen unter Umständen
lohnenswert, etwa ein halbes Jahr nur zu beobachten. Bei sekundären Fällen von membranöser GN steht die Behandlung
der Grunderkrankung im Vordergrund.
Die fokal segmentale Glomerulosklerose ist die zweithäufigste Ursache eines nephrotischen
Syndroms im Erwachsenenalter. Sie wird vielfach als eine Verlaufsform der Minimalläsion angesehen, bei der die
Podozyten in einzelnen Glomeruli, d.h. fokal so schwer geschädigt werden, daß sie teilweise zugrunde gehen und
es segmental zu Vernarbungen kommt. Charakteristischerweise treten eine plötzliche schwere Proteinurie,
Ödembildung und Kreatininanstieg auf.
-
Die Mesangioproliferative GN (IgA-Nephropathie/Nephritis, Morbus Berger) ist eine auf die Nieren beschränkte,
immunvermittelte Krankheit. Hierbei handelt es sich um die im Erwachsenenalter häufigste entzündliche Glomerulopathie,
von der etwa dreimal mehr Männer als Frauen betroffen sind. Aus dem gehäuften familiären Auftreten der Krankheit
|
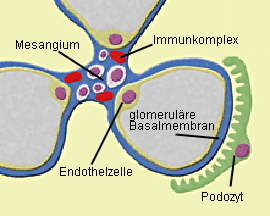
Abb. 2a: Mesangioproliferative GN (IgA-Nephropathie): Die Antikörper – vorwiegend IgA –
werden charakteristischerweise im Mesangium der Glomeruli abgelagert. Diese Ablagerung von IgA führt zur
Proliferation von Mesangiumzellen.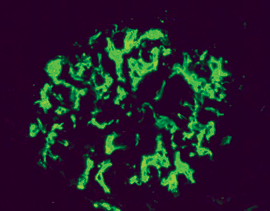 Abb. 2b: Mesangioproliferative GN bei IgA-Nephropathie (Immunfluoreszenz): Die Ablagerung
der Immunkomplexe konzentriert sich auf mesangiale Bereiche.
Abb. 2b: Mesangioproliferative GN bei IgA-Nephropathie (Immunfluoreszenz): Die Ablagerung
der Immunkomplexe konzentriert sich auf mesangiale Bereiche.
|
Worauf die Störung im Immunsystem bei einer IgA-Nephropathie letztlich zurückzuführen ist, läßt sich gegenwärtig nicht beantworten. Die Antikörper – vorwiegend IgA – werden charakteristischerweise im Mesangium der Glomeruli abgelagert – unter Umständen aber auch in anderen Anteilen des Glomerulums (Abb. 2a, b). Im Mesangium führt die Ablagerung von IgA zur Aktivierung der Mesangiumzellen, zur vermehrten Freisetzung von Zytokinen und zur Proliferation von Mesangiumzellen.
Das klinische Bild der IgA-Nephropathie kann sehr stark variieren. Als Minimalbefund findet sich eine geringe persistierende Mikrohämaturie. Dies ist häufig der Fall, wenn die Ablagerung der Immunkomplexe auf das Mesangium beschränkt bleibt. In solchen Fällen ist die Prognose günstig. Werden aber auch IgA-Komplexe im subendothelialen Bereich abgelagert, bewirken sie ein nephritisches Syndrom. Charakteristisch sind rezidivierende Episoden mit Makrohämaturie, die meist von einer Infektion der oberen Luftwege begleitet sind. Ferner besteht neben der Hämaturie meist eine milde Proteinurie. Unter Umständen liegt das klinische und pathologische Bild einer rapid progressiven GN vor. Kommt es aber zu IgA-Ablagerung auch im subepithelialen Bereich, werden die Podozytenfortsätze geschädigt und es entwickelt sich ein nephrotisches Syndrom.
Als therapeutische Maßnahmen kommen bei der IgA-Nephropathie antiinflammatorische, antiproliferative, einer Matrixvermehrung entgegenwirkende und antihypertensive Medikationen zur Anwendung. Das Vorgehen orientiert sich sehr stark am Ausmaß der Proteinurie und damit am Zustand der Nierenfunktion. Grundlegend ist die Blutdruckkontrolle mit dem Zielparameter RR 125/85 mmHg. Bei Patienten mit gut erhaltener Nierenfunktion sind je nach Ausmaß der Proteinurie auch Steroide indiziert, um insbesondere einer Kreatinin-Verschlechterung vorzubeugen. Gelegentlich werden auch Fischöle eingesetzt.
Als ungünstige prognostische Faktoren bei der IgA-Nephropathie gelten eine Proteinurie >1,5 g/die, eine glomeruläre Filtrationsrate <70 ml/min, Bluthochdruck und männliches Geschlecht. Bei einem Teil der Patienten im jungen Erwachsenenalter geht die IgA-Nephropathie in eine chronische Niereninsuffizienz über.
-
Die membranoproliferative GN Typ I ist eine Immunkomplex-Erkrankung mit Komplementaktivierung. Sie tritt
als glomeruläre Begleitreaktion bei systemischem Lupus erythematodes, Hepatitis C/Kryoglobulinämie, HIV und
Leukämien auf. Teilweise (in ca. 20% der Fälle) entwickelt sich das Bild eines akuten nephritischen Syndroms,
während bei 10% bis 20% der Patienten Makrohämaturie-Episoden ähnlich wie bei der IgA-Nephropathie auftreten.
Die Beschreibung als membranoproliferativ beruht auf histopathologischen Befunden. Zum einen ist die Anzahl
der Mesangiumzellen deutlich vermehrt. Andererseits erscheint die Basalmembran der Kapillaren im Lichtmikroskop
deutlich verdickt. Oft ist sie auch als Doppelkontur (tram track) erkennbar, wenn sich Mesangiumzellen zwischen
Endothel und Basalmembran schieben, so daß sich vom Endothel her eine Art zweite glomeruläre Basalmembran ausbildet.
-
Als primäre Erkrankung tritt die akute Glomerulonephritis nach überstandener Infektion mit einer Reihe
unterschiedlicher Erregertypen auf. Unter den Verursachern steht eine Infektion mit beta-hämolysierenden
Streptokokken vom Typ A an erster Stelle. Aber auch andere bakterielle Erreger wie Diplokokken,
Staphylokokken, Treponema pallidum, Corynebakterium bovis, Salmonella typhosa, Brucella suis und
Mykobakterien wurden identifiziert.
Als virale Verursacher akuter Glomerulonephritiden kommen Coxsackievirus, Cytomegalie-Virus, Hepatis-B-Virus, Eppstein-Barr-Virus, Rubella-Virus und Mumpsviren in Frage. Darüber hinaus sind auch mykotische und protozoale Erreger wie Coccidioides immitis, Plasmodium malariae, Plasmodium falciparum, Schistosoma mansoni, Toxoplasma gondi und Trypanosomen als Verursacher akuter Glomerulopathien identifiziert worden.
Die poststreptokokkale GN gilt als Prototyp der akuten postinfektiösen GN. Ihre Häufigkeit hat
sich in den meisten westlichen Ländern über die letzten Jahrzehnte kontinuierlich verringert. Hierfür werden in
erster Linie die verbesserte gesundheitliche Situation mit dem gezielten Einsatz von Antibiotika und gehobene
sozioökonomische Verhältnisse verantwortlich gemacht.
Die akute poststreptokokkale GN tritt beim männlichen Geschlecht etwa doppelt so häufig auf wie beim weiblichen.
|
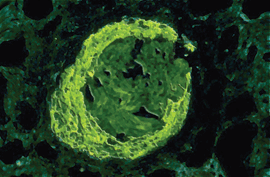
Abb. 3a: Bei der postinfektiösen GN kommt es zur Ablagerung von Immunkomplexen zwischen
Endothel und Basalmembran. Teilweise finden sich auch mesangiale Ablagerungen von IgG und insbesondere
Komplement C3.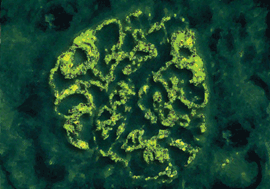 Abb. 3b: Glomerulum bei postinfektiöser GN (C3-Immunfluoreszenz): Die fluoreszierenden granulären Ablagerungen
der Immunkomplexe erscheinen unregelmäßig im Glomerulum verstreut.
Abb. 3b: Glomerulum bei postinfektiöser GN (C3-Immunfluoreszenz): Die fluoreszierenden granulären Ablagerungen
der Immunkomplexe erscheinen unregelmäßig im Glomerulum verstreut.
|
Bei der akuten postinfektiösen GN lagern sich Immunkomplexe endokapillär, d.h. subendothelial zwischen Endothel und glomerulärer Basalmembran ab (Abb. 3a, b). Ferner kommt es zur Komplementaktivierung und Proliferation von Mesangium- und Endothelzellen. Exsudative Prozesse führen zur Infiltration von Leukozyten und Verlegung des Kapillarlumens.
In der Therapie der poststreptokokkalen GN sind Antibiotika indiziert. Bei einer sich verschlechternden Nierenfunktion, werden auch Glukokortikoide eingesetzt.
Im Kindesalter ist die langfristige Prognose bei poststreptokokkaler GN sehr gut. Hingegen sind die Aussichten auf eine vollständige Ausheilung bei den erwachsenen Patienten deutlich weniger günstig insbesondere wenn die Krankheit mit einer deutlichen Beeinträchtigung der Nierenfunktion einsetzt. Im Erwachsenenalter wird auch vermehrt die Ausbildung von Halbmonden beobachtet.
Beim Abklingen der Entzündung kommt es unter Umständen zu einer Verlagerung von Immunkomplexen von einer Seite der glomerulären Basalmembran auf die andere, d.h. von der subendothelialen in die subepitheliale Position. Schädigen diese translozierten Immunkomplexe die Podozyten, kann es in einigen wenigen Fällen auch zur Ausbildung eines nephrotischen Syndroms kommen.
-
Der pathologische Nachweis fibrinoider Nekrosen und Halbmondbildung (Abb. 4) in mehr als 50%
der Glomeruli ist charakteristisch für die rapid progressive GN. Diese subakute bzw. akute Nierenerkrankung führt
nach abruptem Beginn zu einem rapid progressiven Kreatininanstieg und zum raschen Verlust der Nierenfunktion.
Innerhalb von nur drei Monaten sinkt die glomeruläre Filtrationsrate auf unter 50%. Die Krankheit verursacht
in der Regel ein nephritisches Syndrom. Im wesentlichen handelt es sich um sekundäre Nephropathien, die als
Begleitreaktion systemischer Krankheiten auftreten. Man unterscheidet drei Formen der rapid progressiven GN:
I Anti-Basalmembran-GN
II Immunkomplex-GN
III Pauci-Immun-GN
Ihre komplexe Diagnostik beruht auf serologischen Parametern, der Beurteilung des klinischen Bildes und der histopathologischen Untersuchung von Nierenbiopsien.
Beim seltenen Goodpasture-Syndrom (rapid progressive GN Typ I) bildet das Immunsystem Autoantikörper gegen die 3-Kette des Kollagens Typ IV, das sowohl in der glomerulären Basalmembran (GBM) als auch in der Basalmembran
|
Abb. 4: Halbmond (Immunfluoreszenz): Als Folge extrakapillärer Zellproliferation und
Fibrinansammlung in der Bowmanschen Kapsel wird das Kapillarkonvolut verdrängt. 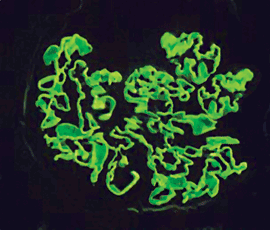 Abb. 5: Goodpasture-Syndrom (Immunfluoresz): Die glomeruläre Basalmembran ist
gleichmäßig mit fluoreszierenden Anti-IgG-Antikörpern besetzt.
Abb. 5: Goodpasture-Syndrom (Immunfluoresz): Die glomeruläre Basalmembran ist
gleichmäßig mit fluoreszierenden Anti-IgG-Antikörpern besetzt.
|
Das klinische Bild der rapid progressiven GN beim Goodpasture-Syndrom ist durch einen rasch eintretenden Funktionsverlust der Nieren und durch Lungenblutungen gekennzeichnet. Letztere können unter Umständen lebensbedrohend sein. In der Therapie beim Goodpasture-
Von einer Anti-GBM-Krankheit wird gesprochen, wenn nur die Nieren involviert sind.
Bei der rapid progressiven GN Typ II handelt es sich um äußerst schwere Immunkomplexerkrankungen. Dieses Krankheitsbild kann unter anderem als fulminante Form der postinfektiösen GN auftreten. Ferner wird es bei der IgA-Nephropathie, der membranoproliferativen GN und bei Henoch-Schönlein-Purpura beobachtet.
Bei der rapid progressiven GN Typ III finden sich so gut wie keine Immunablagerungen (Pauci-Immun-GN).
Dieser GN-Typ entwickelt sich im Zusammenhang mit Vaskulitiden. Charakteristisch ist das Auftreten von Autoantikörpern,
die gegen Antigene in den Granula der segmentkernigen Leukozyten gerichtet sind. Diese Anti-Neutrophil-
Die generalisierte Wegenersche Granulomatose wird durch die Trias aus nekrotisierenden Granulomen
im oberen oder unteren Respirationstrakt, eine generalisierte nekrotisierende Vaskulitis sowohl der Arterien als auch
der Venen und eine nekrotisierende Glomerulonephritis definiert. Durch die Nierenbeteiligung kommt es zu einem subakuten
Kreatininanstieg, zu mäßiger Proteinurie und Mikrohämaturie. Im nephritischen Sediment findet man Erythrozytenzylinder
und Akanthozyten. Bei der pathologischen Untersuchung lassen sich c-ANCA nachweisen.
Die mikroskopische Polyangitis ist eine meist p-ANCA-positive Vaskulitis mit rapid progressiver GN.
Gelegentlich kommt sie auch als ein auf die Nieren beschränkter Krankheitsprozeß vor (nekrotisierende Halbmondnephritis).
Die Prognose der Wegenerschen Granulomatose hat sich mit Einführung der Kombinationstherapie aus Steroiden und
Cyclophosphamid erheblich gebessert.
© 2003-2026 pro-anima medizin medien
–
impressum
–
mediadaten
–
konzeption
–
datenschutz